Samuel Alizon, chercheur en biologie de l’évolution au sein du Laboratoire des Maladies infectieuses et vecteurs : écologie, génétique, évolution et contrôle (MIVEGEC) répond à nos questions. L’équipe ETE, dont il fait partie, est rattachée à ce laboratoire interdisciplinaire où l’on étudie les maladies infectieuses dans une perspective que l’on appelle One Health, de Santé Globale, avec des approches qui vont du terrain jusqu’au laboratoire, et à la modélisation mathématique et statistique.
En quelques mots, quel est votre parcours ?
Je suis biologiste de formation et me suis spécialisé assez vite dans la modélisation autour de questions d’épidémiologie et de biologie de l’évolution. Aujourd’hui je travaille, comme toute l’équipe ETE, à l’interface entre biologie et modélisation mathématique et statistique.
Spécialiste en modélisation des maladies infectieuses, qu’est-ce que c’est ?
Dès que l’on regarde ou représente des données, il y a de la modélisation, un modèle sous-jacent. Nous utilisons trois grands types de modèles dans l’équipe : les modèles statistiques qui sont très adaptés pour analyser des données, les modèles computationnels, qui consistent à faire des simulations informatiques les modèles mathématiques, qui utilisent des représentations plus simplifiées de la réalité, mais ont des puissances d’analyse et de prévision supérieures. Un des exemples dont on parle beaucoup en ce moment est le R0, le nombre moyen de personnes qu’infecte quelqu’un au cours de son infection dans une population entièrement sensible et où il n’y a pas de mesures barrières. Si R0>1 l’épidémie peut croître alors que sinon l’épidémie est en décroissance. Ce nombre peut être exprimé à l’aide d’un modèle mathématique, simulé à l’aide d’un modèle informatique ou estimé à partir des données à l’aide d’un modèle statistique. Notre équipe a d’ailleurs été la première à publier une estimation du R0 de l’épidémie de SARS-CoV-2 en France, à la mi-mars.
Les modèles nous aident à décrire, à comprendre et aussi à prévoir le comportement de l’épidémie. Plus on travaille à une échelle large et homogène (par exemple la France), plus on peut faire des prévisions déterministes car la loi des grands nombres s’applique. À l’inverse, plus l’échelle est fine (un quartier d’une ville), plus les modèles mathématiques montrent leurs limites parce que vous avez beaucoup de facteurs qui vont entrer en compte. Les modèles numériques peuvent alors être plus adaptés car on peut y inclure de nombreux détails. Bref, il y a toujours un compromis entre précision et généralisme. De plus, plus on se projette sur le long terme, moins on peut prévoir. Pour l’épidémie actuelle, au-delà de 2 semaines il est impossible d’avoir beaucoup de certitudes.
Durant cette période pandémique vous avez déployé plusieurs outils, Rt2 et COVIDici, en quoi consistent-ils ? A quel(s) besoin(s) répondent-ils? A qui s’adressent-t-ils?
Les modèles nous aident à décrire, à comprendre et aussi à prévoir le comportement de l'épidémie"
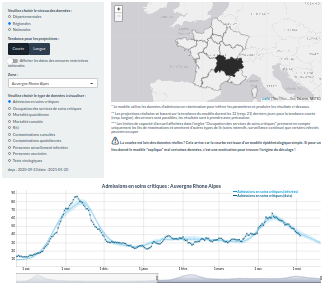
Rt2 est un outil de modélisation statistique, qui analyse les données de manière relativement brute, ou heuristique. Cet outil permet à l’utilisateur de choisir, selon un lieu donné, à une échelle donnée (pays, département, région), une visualisation et une analyse de données d’incidence de plusieurs types : nouveaux cas dépistés, nouvelles hospitalisations, nouvelles admissions en réanimation et nouveaux décès. Rt2 calcule ce que l’on appelle le nombre de reproduction temporel, qui ressemble au R0 mais estimé à un moment donné, donc dans une population plus ou moins immunisée et où des mesures de contrôle peuvent être en place .
Au tout début, on ne publiait nos estimations que ponctuellement. L’interface graphique est venue ensuite, grâce au package Shiny du langage programmation R, qui permet d’avoir une interface utilisateur avec un choix du pays et du type de données. Shiny permet de passer assez facilement des scripts avec lesquels on travaille à une interface accessible pour un public plus large. Au début, ce sont des collègues du serveur de bioinformatique de l’IRD à Montpellier qui nous ont aidé pour l’hébergement web. Puis nous nous sommes tournés vers l’IFB pour une solution plus pérenne et être recensé dans les initiatives prises au sujet de la Covid au niveau national.
Nos outils de visualisation sont à destination du grand public, afin que les gens puissent aller voir chaque jour dans un département donné quel est l’état de l’épidémie. Le logiciel automatise les calculs et permet de la faire à différentes échelles. On a des retours de médecins en infectiologie au CHU de Montpellier, qui utilisent ces deux outils quotidiennement parce qu’ils donnent une visualisation mais aussi des tendances à un niveau départemental. Depuis début 2021, on compte plus de 800 visiteurs uniques pour Rt2 et plus de 12 000 pour COVIDici. Comme pour les autres sites qui font de la représentation de données, il est clair que cela ne serait pas possible sans le partage des données réalisé par les Autorités Régionales de Santé (ARS) et Santé Publique France.
La première version du site Rt2 était en ligne vers la mi-avril 2020. COVIDici a été lancé beaucoup plus récemment, en février 2021. Il fonctionne aussi à l’échelle nationale, régionale et départementale, et représente différents types de données en se basant sur les admissions en réanimation. Son originalité est qu’en plus de permettre une visualisation des données, il montre aussi les résultats d’estimations associées à un modèle mathématique sous-jacent qui capture la dynamique épidémique. Grâce à ces modèle sous-jacent, qui prend en compte la vaccination depuis mars, on peut tenter d’estimer des valeurs non mesurables comme par exemple le nombre de personnes porteuses de l’infection à un moment donné. Un des autres intérêts d’avoir un modèle sous-jacent est que l’on peut faire des prévisions sur les tendances sur la semaine à venir en faisant l’hypothèse que la tendance observée au cours des deux dernières semaines va se prolonger.
En termes de collecte des données, comment procédez-vous ?
En mars l’année dernière il fallait presque aller à la pêche aux données. Depuis avril 2020, le site Data.gouv.fr met à jour quotidiennement des indicateurs hospitaliers pour la France grâce à Santé Publique France. Les deux scripts, Rt2 et COVIDici, vont chaque soir récupérer les nouvelles données sur ce site avant de recalculer les estimations de paramètres du modèle sous-jacent et de mettre à jour les valeurs et graphiques.
Pour Rt2, le calcul se fait à chaque requête d’utilisateur mais on récupère en plus des données qui sont compilées par le site Ourworldindata qui est un site qui regroupe des données au niveau mondial, parce que l’outil Rt2 permet aussi de calculer le nombre de reproductions dans différents pays.
La singularité de vos outils vient de leur puissance d’estimation, doit-on comprendre que les besoins sont essentiellement au niveau prédictif dans un monde scientifique, politique et global en crise sanitaire ?
Notre équipe a commencé à travailler relativement tardivement en fait sur l’épidémie quand on y pense, aux alentours du 9-10 mars 2020. On pensait, peut-être naïvement, que beaucoup d’équipes d’épidémiologie en France allaient faire ce travail d’estimation de paramètres, comme on le voyait alors en Angleterre. En matière de modélisation en épidémiologie, il est important d’avoir plusieurs équipes qui travaillent de concert En effet, chaque équipe va utiliser des modèles et des hypothèses différents pour répondre à une même question, et la confrontation des modèles vous en apprend toujours énormément. Si tous s’accordent sur des prévisions identiques, vous êtes assez confiants. S’ils divergent fortement, vous allez en apprendre beaucoup parce qu’en analysant les hypothèses de chacuns vous allez pouvoir expliquer pourquoi tel modèle génère des prévisions différentes.
Cela permet d’identifier les hypothèses clés et les données manquantes pour trancher. Et vu le peu d'équipes qui travaillaient dessus, il y avait vraiment un besoin de modélisation.
Au début, on faisait ces estimations pour nous, on les refaisait à peu près une fois par semaine, et on s’est aperçu qu’il y avait une demande assez forte. Par exemple, des structures locales, des CHU, avaient des retours uniquement quand le conseil scientifique publiait des analyses, c’est-à-dire de manière irrégulière et au niveau régional, pas départemental. COVIDici répond à ce besoin même si les tendances au niveau départemental sont à prendre avec beaucoup de précautions car on est loin de la “loi des grands nombres”.
Pour l'épidémie actuelle, au-delà de 2 semaines il est impossible d'avoir beaucoup de certitudes"
La crise actuelle pose la question de la coordination nationale, quid de la France ?
L’épidémiologie mathématique était marginale en France avant la pandémie. C’était une discipline un peu négligée. Par exemple, bien que l’agence Santé Publique France soit intéressée par la modélisation et collabore avec quelques modélisateurs, elle n’a pas de département de modélisation. On partait aussi de loin pour tout ce qui était la centralisation et le partage des données. Nos approches de modélisation statistique ont par exemple été bloquées jusque fin avril 2020 en l’absence de données hospitalières.
Aujourd’hui, on ne dispose toujours pas de données de suivi de contact qui permettraient d’estimer un intervalle sériel (le temps entre deux infections) en France. Cette carence nous rappelle que la récolte, la centralisation et le stockage des données ont un coût. Clairement, on paye le prix du peu d’investissements en santé publique et en recherche en France au depuis plus de 20 ans.
Racontez-nous la collaboration avec l’IFB, comment s’est-elle déroulée ? En quoi a constitué pour vous le support de l’IFB : aide, conseil, hébergement ?
Concrètement, on avait besoin d’un hébergement pour un accès public. La difficulté était qu’il ne s’agit pas d’un site web fixe classique, car en tâche de fond il y a des calculs à effectuer. Il fallait articuler à la fois l’accès au site, et toute la partie calcul derrière ; et c’est ce que permet entre autres ce serveur Shiny. On a pris contact fin décembre avec l’IFB, et un mois après le site était en ligne, hébergé sur le Cloud de l’IFB-core à Lyon.
Nos outils de visualisation sont à destination du grand public, afin que les gens puissent aller voir chaque jour dans un département donné quel est l'état de l'épidémie"
Autant pour Rt2 c’était quelque chose de très simple puisque le script Shiny était déjà en place et on l’a repris tel quel. Autant pour COVIDici c’était plus technique. Sur COVIDici, toutes les valeurs sont d’abord calculées et ensuite il y a le site web avec le serveur Shiny qui permet de visualiser et à l’utilisateur de choisir ce qu’il veut voir.
L’avantage de cette interface Shiny c’est de passer facilement des scripts de recherche à des interfaces plus accessibles au grand public. Il a fallu coupler à la fois la partie calcul et la partie représentation. Pour nous ça a été vraiment très confortable de passer par l’IFB, pour le suivi, mais aussi la facilité et la souplesse des mises à jour. Christophe Blanchet nous a aussi beaucoup aidé au niveau des containers, pour faire des scripts qui permettent d’automatiser certaines étapes. Il a aussi pris en charge de nombreux aspects : la paramétrisation des machines ou encore tout ce qui concerne les calculs qui se font la nuit en tâche de fond.
D’autres acteurs ont-ils pris part à ces projets ?
L’équipe i-Trop de l’IRD Montpellier nous hébergeait au début et nous a mis en contact avec l’IFB. Ce travail a aussi été possible grâce à la région Occitanie, qui a été la seule institution à nous soutenir (nous sommes sans nouvelles de nos tutelles et tous nos autres projets de recherche ont été refusés).
Cela a permis de payer le salaire de l’ingénieur qui a écrit les scripts pour COVIDici notamment, et qui aujourd’hui fait les mises-à-jour.
Avez-vous d’autres besoins de support pour ces outils qui ne cessent d’évoluer en fonction de l’épidémie ?
Je ne sais pas si les gens se rendent compte à quel point on travaille avec des bouts de ficelle... L’équipe a pourtant joué le jeu avec 3 projets ANR déposés (un sur la phylodyamique, un avec le CHU pour estimer les infections secondaires dans les foyers de l’Hérault et un sur la modélisation de la propagation spatiale) et un projet ANRS sur la modélisation de l’épidémie en Afrique. Finalement, tous ont été refusés et seule la région a accepté de financer l’un des quatre qui était sur liste complémentaire. Du côté de nos tutelles (CNRS, IRD, Université de Montpellier), aucun soutien. En fait, nous avons surtout perdu de la place de bureau en recrutant un ingénieur. Et oui nous aimerions faire plein d’autres choses sur l’épidémie de COVID-19 : des analyses statistiques en temps réel sur les données de tests PCR, des analyses des épidémies dans les EHPADs, déposer des projets européens...
Le plus frustrant est probablement de ne pas avoir le temps d’analyser les séquences génétiques, par exemple avec des méthodes de phylodynamique. Cela permettrait par exemple de comparer la vitesse de propagation des différentes lignées virales. Ces données de génomes viraux sont maintenant partagées de manière assez rapide via la plateforme GISAID, ce qui n’était pas le cas fin mars 2020, où il n’y en avait que 200 pour la France. Aujourd’hui on est à plusieurs milliers, mais les analyses sont faites par des équipes suisses ou anglaises qui sont bien financées. Nous, nous sommes spectateurs car à un moment les journées durent 24h et toute l’équipe est en surcharge de travail, puisque la plupart des autres projets non-COVID continuent aussi.
Vous parlez souvent de l’importance d’un processus co-évolutif, entre politique de santé publique et évolution virale , ceci est-il effectif dans les prises de décisions françaises ?
On a parlé de la modélisation mathématique qui était marginale en France en santé publique. La biologie de l’évolution l’était encore plus (et pour ce qui est de la modélisation en évolution, inutile d’en parler). Clairement, la mise en place d’un conseil scientifique a été quelque chose de très positif, même s’il est malheureusement maintenant marginalisé par les autorités qui ne goûtent pas ses analyses.
D’ailleurs, je n’aurais pas parié sur la présence d’un modélisateur dans un tel conseil il y a deux ans. Selon moi, cela a beaucoup aidé à la pertinence de leurs analyses. En revanche, que la biologie de l’évolution n’y soit pas représentée est dommage car cela aiderait à avoir une vision de long terme.
Avec les variants est arrivée la thématique de la mutation, de la variation des virus, quid du dilemme entre le hasard des variations virales et l’apport des outils de prédiction ?
A la base de l’évolution, il y a toujours la mutation qui est un phénomène dit stochastique, qui dépend du hasard. Vous ne pouvez pas prévoir où aura lieu la prochaine mutation. Cependant avec les virus, ou les microbes en général, les tailles de populations sont tellement grandes, que ce hasard est limité. Quand une personne est infectée, son organisme va produire des millions de virus, dont un certain nombre de mutants. Si 100.000 personnes sont infectées, il est probable que toutes les mutations possibles se produisent au moins une fois. Donc la loi des grands nombres finit finalement par compenser ce côté aléatoire de la mutation. Comme je le détaille dans mon livre, il faut donc voir le virus non pas comme une cible fixe, mais une cible mouvante. De plus, les mesures prises influent potentiellement sur l’évolution virale.
J’ai été vraiment surpris jusqu’à la fin 2020 de l’évolution virale. On voyait que les virus qui se propageaient en France n'étaient pas issus des mêmes lignées que ceux qui se propageaient en Asie, ou en Amérique du Sud. Toutefois, la plupart des mutations associées aux lignées étaient neutres. Autrement dit, les virus différaient les uns des autres, mais c’était sans conséquence sur l’infection elle-même. Depuis la fin 2020 c’est différent. On a ce qu’on appelle les variants préoccupants - on parle d’habitude plutôt de souches. Ces virus ont plus de mutations que la moyenne, mais surtout, les infections qu’ils causent sont différentes. Pour le variant V1, qui a d’abord été détecté en Angleterre, il a été montré qu’il est plus contagieux. En France aussi notre équipe a estimé une hausse de 40% de sa contagiosité par rapport aux lignées précédentes (aussi appelées souches sauvages). Les premières données d’Angleterre semblent montrer qu’il est aussi plus virulent, c’est-à-dire que les personnes qui sont infectées par ce variant ont un plus grand risque de décéder. Deux autres variants préoccupants sont apparus, V2 et V3, en Afrique du Sud et au Brésil, qui semblent aussi être plus contagieux, quoique dans une moindre mesure que V1. Avec les mêmes méthodes, nous estimons une hausse de contagiosité de V2 par rapport aux souches sauvages de l’ordre de 25%.
Sur la virulence on n’a pas encore de donnée, mais il semblerait que ces deux variants V2 et V3 sont capables de contourner la réponse immunitaire, c’est-à-dire la réponse acquise après une infection naturelle.
Les personnes infectées pendant la première vague par une souche ancestrale, sont à priori protégées contre une réinfection, même par le variant V1; en revanche, elles sont exposées à une réinfection par les variants V2 ou V3.
L’émergence des variants V2 et V3 est assez cohérente avec le déroulement de l’épidémie. Si on se place du point de vue d’un virus, son but est d’infecter le plus d’hôtes possible, sauf qu’à un moment, beaucoup de gens sont immunisés. Cette immunité agit comme une pression de sélection, de telle sorte que les virus qui sont capables de contourner cette immunité sont extrêmement favorisés. Certes la mutation arrive au hasard, mais le fait que le variant V3 ait été détecté au Brésil, soit dans un pays où il y avait beaucoup de gens immunisés, est assez logique.
Ce sont des outils qui fonctionnent à l'échelle nationale, régionale et départementale"
Pour ce qui est de l’immunité vaccinale, de son efficacité, il est en théorie possible que les mutations des virus aient des effets sur l’efficacité des vaccins. Si on prend un virus comme le VIH, c’est pour ça qu’on n’a toujours pas de vaccin contre l’infection, parce que le virus évolue tellement rapidement qu’il contourne l’immunité vaccinale. Dans le cas du SARS-Cov-2, pour le moment les nouvelles sont plutôt bonnes, enfin surtout pour les vaccins ARN, qui semblent relativement robustes aux nouvelles souches. Mais, en théorie, on est toujours à la merci d’un virus qui échappe à l’immunité vaccinale.
Ces variations sont-elles infinies ?
En vaccinant, vous exercez une pression de sélection qui favorise les souches qui échappent au vaccin. C’est assez mécanique en fait. Mais en vaccinant, vous diminuez aussi la circulation du virus et donc le nombre de nouveaux mutants. C’est un peu pour ça que le Conseil Scientifique avait parlé d’une course contre la montre entre les vaccins et les variants. Malheureusement, si vous vaccinez une petite fraction de la population en laissant circuler l’épidémie, là ce n’est pas terrible parce que vous avez une génération continuelle de mutants et une pression de sélection.
Une précision importante est qu’un des avantages des vaccins ARN, c’est qu’on peut, en théorie, mettre à jour relativement rapidement la formule vaccinale car le vaccin cible un motif du génome du virus. D’une certaine manière on peut avoir des vaccins qui co évoluent avec les virus beaucoup plus rapidement. Autre nouvelle positive, les vaccins ARN semblent pour le moment bien résister à l’évolution virale, même sans mise à jour.
Un avenir inéluctablement partagé avec des maladies infectieuses, voir des pandémies, c’est ce qui nous attends ?
C’est quelque chose dont j’ai toujours été convaincu. Il y a eu cette illusion à la fin des années 60 qu’on allait pouvoir éradiquer toutes les maladies infectieuses, parce qu’on avait éradiqué la variole et que l’on découvrait sans cesse de nouveaux antibiotiques. Tout ça a été complètement remis en cause fin des années 80-début 90, avec la pandémie de VIH, et aussi la généralisation de l’antibiorésistance. Finalement l’émergence du SARS-CoV 2 est un peu le coup de grâce pour cette théorie de l’optimisme qui voudrait croire que l’on peut éradiquer toutes les maladies infectieuses. L’enjeu aujourd’hui est d’abord d’en être conscient.
Les microbes font partie de notre environnement, et c’est là où il faut essayer d’anticiper, et d’éviter ce que l’on appelle une course aux armements (...) "
Il faut aussi préciser que cette co-évolution ne se place pas tant au niveau de l’évolution humaine qu’au niveau des politiques de santé, et politiques publiques en général. L’exemple classique c’est l’utilisation des antibiotiques : plus vous utilisez d’antibiotique plus l’antibiorésistance évolue et se généralise. C’est un peu similaire pour l’émergence de la pandémie de SARS-CoV 2 où l’on sait qu’il y a des facteurs de risques qui augmentent ce qu’on appelle des zoonoses, c’est-à-dire le passage de virus depuis des populations naturelles, animales, vers la population humaine. Tout ce qui va mettre en contact la population humaine avec ces populations sauvages, l’urbanisation, la déforestation, etc., sont des facteurs de risque.
Les microbes font partie de notre environnement, et c’est là où il faut essayer d’anticiper, et d’éviter ce que l’on appelle une course aux armements, c’est-à-dire des phénomènes qui sélectionnent des souches microbiennes toujours plus virulentes, pour au contraire essayer d’aller vers des approches plutôt de coexistence pacifique, qui permettent justement de contrer l’évolution des souches virulentes.